

多重置换扩增(MDA)是现阶段最常用的单细胞全基因组扩增方法,与第三代测序(TGS)相结合,可以对长度超过20kb的扩增片段进行测序。然而,MDA扩增过程中错误启动会产生嵌合序列(测序数据中表现为结构错误)是不可避免的问题,在已有的使用MDA制备模板的所有二代测序(NGS)数据中嵌合序列的比例在1%至15%范围内。考虑TGS更长的读长,其中的嵌合序列的比例预计会更高,影响TGS数据利用率,增加数据分析的难度,需要评估影响。
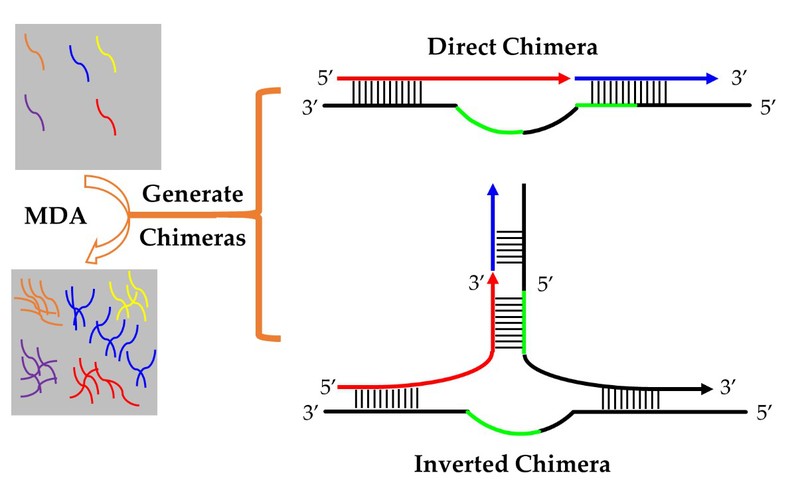
图1. 嵌合序列的形成机制
本研究构建了3rd-ChimeraMiner,首次利用PacBio单分子测序平台对人类TGS测序数据中MDA产生的嵌合序列进行了系统地探索。结果表明,MDA中的错误启动事件发生频率比之前在短读测序数据中认识到的更高,嵌合事件随着扩增不断累积,单细胞级别数据中嵌合比例高达78%。通过将嵌合序列恢复到原始结构,可在召回大部分真实结构变异的同时(78.23%)去除大部分假阳性结构变异(94.11%),使得在单细胞TGS测序数据中检测结构变异成为可能。
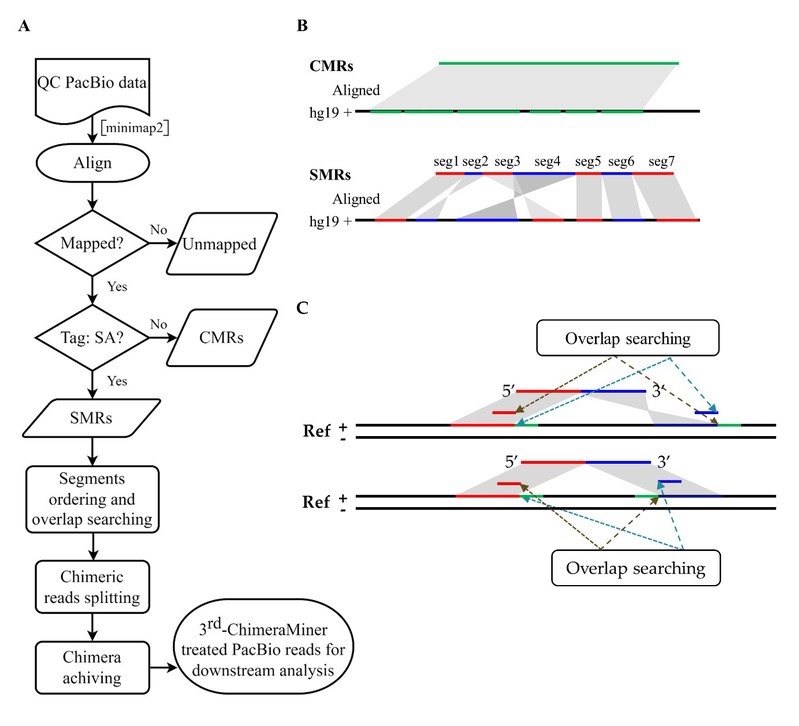
图2. 3rd-ChimeraMiner的概述图
这是一项原理验证研究,结合MDA和TGS,对嵌合序列进行识别和再利用,实现TGS在单细胞水平研究中的应用。
论文链接:https://doi.org/10.1093/bib/bbad275.
分析工具链接:https://github.com/dulunar/3rdChimeraMiner.

